


Атаксия-телеангиэктазия (АТ), исторически известная как синдром Луи-Бар, в медицинских учебниках классически описывается как тяжелое, быстро прогрессирующее нейродегенеративное заболевание детского возраста. Генетики связывают его с триадой симптомов: нарастающей мозжечковой атаксией (расстройством координации), сосудистыми «звёздочками» (телеангиэктазиями) и выраженным иммунодефицитом.
Однако развитие таргетного секвенирования (NGS) показало, что клиническая реальность шире учебников. Существует вариантный (или атипичный) тип атаксии-телеангиэктазии, который долгое время оставался «невидимкой» для клиницистов. Главная его особенность — относительно мягкое течение, обусловленное частичной сохранностью функции ключевого белка клеточного выживания.
Генетический фундамент: Сила «остаточной активности»
В основе классического синдрома Луи-Бар лежат мутации в гене ATM (Ataxia Telangiectasia Mutated), расположенном на соответствующей хромосоме. Этот ген кодирует одноименный белок-киназу ATM — «главного диспетчера» ответа клетки на повреждения ДНК. Когда в ДНК возникают опасные двунитевые разрывы (например, под воздействием радиации или свободных радикалов), именно белок ATM активирует системы восстановления (репарации) или запускает апоптоз (самоуничтожение) поврежденной клетки.
- При классической форме АТ обе копии гена несут так называемые null-мутации (нонсенс-мутации, сдвиг рамки считывания). Белок ATM либо не синтезируется вообще, либо полностью лишен киназной активности. Клетки (особенно клетки Пуркинье в мозжечке) беззащитны перед хаосом мутаций и быстро гибнут.
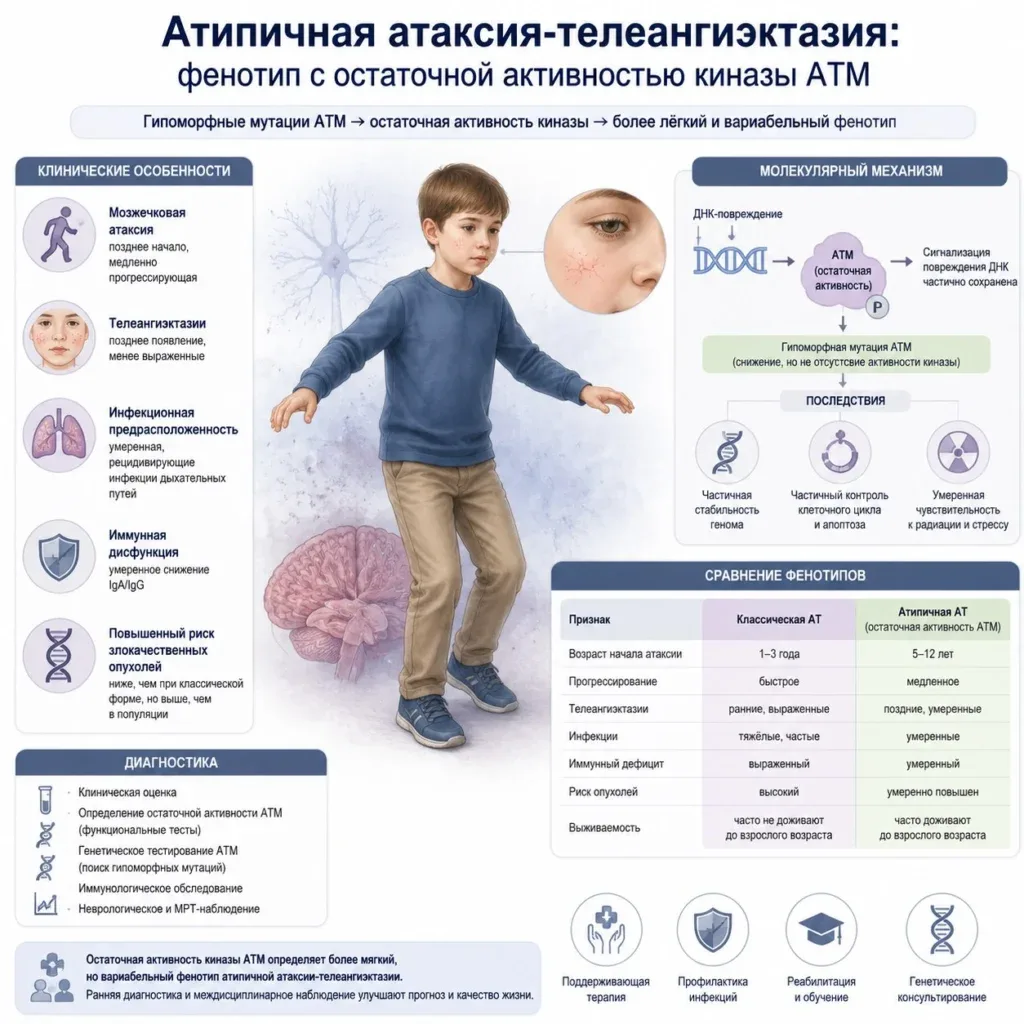
- При вариантном типе АТ пациент наследует хотя бы одну «мягкую» мутацию (например, миссенс-мутацию или мутацию сплайсинга). Это приводит к тому, что в организме сохраняется остаточная функция белка ATM (от 1% до 20% от нормы). Даже такого минимального его количества достаточно, чтобы частично защитить ДНК от повреждений, что кардинально меняет сценарий развития болезни.
Клинический портрет атипичной формы
Вариантный тип АТ характеризуется чрезвычайным клиническим полиморфизмом. Заболевание может манифестировать не в раннем детстве (как классическая форма в 1–2 года), а в подростковом или даже взрослом возрасте (описаны случаи дебюта после 30-40 лет).
1. Неврологические особенности: Сдвиг в сторону дистонии
Если при классическом синдроме Луи-Бар доминирует чистая и быстро прогрессирующая мозжечковая атаксия, то при вариантном типе на первый план часто выходят экстрапирамидные нарушения:
- Дистония (непроизвольные мышечные сокращения, приводящие к спазмам и аномальным позам).
- Хореоатетоз (быстрые, отрывистые или медленные извивающиеся движения).
- Миоклонии (кратковременные подёргивания мышц).
- Тремор (как интенционный, так и тремор покоя, напоминающий паркинсонизм).
Мозжечковая симптоматика развивается значительно медленнее. Пациенты с вариантным типом могут сохранять способность к самостоятельному передвижению до глубокой зрелости, в то время как при классической форме инвалидное кресло требуется уже к 10–12 годам.
2. Соматические маркеры: Маскировка симптомов
- Телеангиэктазии (сосудистые сетки): При атипичной форме они могут появляться очень поздно (к 20–30 годам), быть единичными (только на склерах глаз или в ушных раковинах) либо отсутствовать вовсе. Это одна из главных причин, почему диагноз не выставляется годами.
- Иммунный статус: В отличие от классической формы, сопровождающейся тяжелыми синусопульмональными инфекциями из-за дефицита IgA и IgG2, у пациентов с вариантным типом иммунная система чаще всего относительно сохранна. Риск частых пневмоний и гнойных инфекций существенно ниже.
- Онкологическая настороженность: Риск развития злокачественных новообразований (особенно лимфом и лейкозов) остается повышенным по сравнению с общей популяцией, поскольку репарация ДНК все же скомпрометирована. Однако пик онкозаболеваний смещается на зрелый возраст, и частота эпителиальных опухолей (рак груди, желудка) возрастает.
Проблема диагностики: Почему их сложно найти?
Диагностика вариантного типа АТ — серьезный вызов для неврологов. Из-за отсутствия телеангиэктазий и иммунодефицита таких пациентов годами ведут с ошибочными диагнозами: детский церебральный паралич (ДЦП), идиопатическая торсионная дистония, болезнь Гентингтона или наследственная спастическая параплегия.
Современный алгоритм верификации атипичной АТ включает:
- Генетическое тестирование (NGS): Полноэкзомное секвенирование или панели «Наследственные атаксии» позволяют выявить патогенные варианты в гене ATM.
- Скрининг уровня альфа-фетопротеина (АФП) в сыворотке крови: Это важнейший биомаркер. Как при классической, так и при вариантной форме АТ уровень АФП повышен у 90-95% пациентов. Если у пациента с неясной дистонией или атаксией повышен АФП — это прямой повод искать мутации в гене ATM.
- Вестерн-блоттинг и оценка функциональной активности ATM: Исследование культуры клеток (лимфобластов или фибробластов) пациента. Метод позволяет физически подтвердить наличие белка ATM (пусть и в сниженном количестве) и оценить его способность к фосфорилированию мишеней после радиационного облучения.
Стратегия ведения и терапия
На сегодняшний день радикального лечения (генной терапии), способного исправить мутацию в гене ATM, не существует. Однако терапия вариантной формы имеет свои фундаментальные особенности:
Критически важно: Радиационная безопасность Даже при сохранности части функций белка ATM, клетки пациентов остаются гиперчувствительными к ионизирующему излучению. Пациентам с вариантным типом АТ строго противопоказаны рутинные рентгенологические исследования и КТ (компьютерная томография) без жизненно важных показаний. Для визуализации головного мозга должна использоваться исключительно МРТ.
- Симптоматический контроль двигательных расстройств: Активно используются препараты для коррекции дистонии и гиперкинезов
- Регулярный онкоскрининг: Учитывая пожизненный риск канцерогенеза, пациентам показана диспансеризация с акцентом на раннее выявление гематологических и солидных опухолей
- Нейрореабилитация: Поддержание двигательной активности, ЛФК, занятия с логопедом (для коррекции дизартрии, если она развивается) играют ключевую роль в долгосрочном прогнозе
Заключение
Вариантный тип атаксии-телеангиэктазии меняет наше представление о синдроме Луи-Бар как о исключительно детской фатальной патологии. Остаточная активность белка ATM размывает жесткие рамки классического фенотипа, превращая болезнь в медленно текущее неврологическое расстройство, манифестирующее в более взрослом возрасте. Своевременное распознавание этой формы позволяет избежать неоправданных обследований, защитить пациента от опасной лучевой нагрузки и выстроить адекватную долгосрочную стратегию ведения.